METHODS OF ANALYSIS
Foreign Organic Matter
Test Sample—
Unless otherwise specified in the individual monograph,weigh the following quantities of the laboratory sample,taking care that the withdrawn portion is representative (quartering if necessary):
| Roots,rhizomes,bark,and herbs | 500g |
| Leaves,flowers,seeds,and fruit | 250g |
| Cut vegetable drugs (average weight of the pieces is less than 0.5g) |
50g |
Spread the sample out in a thin layer,and separate the foreign organic matter by hand as completely as possible.Weigh it,and determine the percentage of foreign organic matter in the weight of drug taken.
Total Ash
Accurately weigh a quantity of the Test Sample,representing 2to 4g of the air-dried material,in a tared crucible,and incinerate,gently at first,and gradually increase the temperature to 675±25 ,until free from carbon,and determine the weight of the ash.If a carbon-free ash cannot be obtained in this way,extract the charred mass with hot water,collect the insoluble residue on an ashless filter paper,incinerate the residue and filter paper until the ash is white or nearly so,then add the filtrate,evaporate it to dryness,and heat the whole to a temperature of 675±25.If a carbon-free ash cannot be obtained in this way,cool the crucible,add 15mLof alcohol,break up the ash with a glass rod,burn off the alcohol,and again heat the whole to a temperature of 675±25.Cool in a desiccator,weigh the ash,and calculate the percentage of total ash from the weight of the drug taken.
,until free from carbon,and determine the weight of the ash.If a carbon-free ash cannot be obtained in this way,extract the charred mass with hot water,collect the insoluble residue on an ashless filter paper,incinerate the residue and filter paper until the ash is white or nearly so,then add the filtrate,evaporate it to dryness,and heat the whole to a temperature of 675±25.If a carbon-free ash cannot be obtained in this way,cool the crucible,add 15mLof alcohol,break up the ash with a glass rod,burn off the alcohol,and again heat the whole to a temperature of 675±25.Cool in a desiccator,weigh the ash,and calculate the percentage of total ash from the weight of the drug taken.
Acid-Insoluble Ash
Boil the ash obtained as directed under Total Ash,above,with 25mLof 3Nhydrochloric acid for 5minutes,collect the insoluble matter on a tared filtering crucible or ashless filter,wash with hot water,ignite,and weigh.Determine the percentage of acid-insoluble ash calculated from the weight of drug taken.
Water-Soluble Ash
Boil the ash obtained as directed for Total Ashwith 25mLof water for 5minutes.Collect the insoluble matter in a sintered-glass crucible or on an ashless filter paper.Wash with hot water,and ignite for 15minutes at a temperature not exceeding 450.Subtract the weight of this residue,in mg,obtained under Total Ash,and calculate the percentage of water-soluble ash with reference to the weight of sample as determined under Total Ash.
Alcohol-Soluble Extractives
Method 1(hot extraction method)—
Transfer about 4g of air-dried,coarsely powdered material,accurately weighed,to a glass-stoppered conical flask.Add 100mLof alcohol,and weigh the flask.Shake,and allow to stand for 1hour.Attach a reflux condenser to the flask,and boil gently for 1hour,cool,and weigh.Readjust to the original weight with alcohol.Shake,and filter rapidly through a dry filter.Transfer 25mLof the filtrate to a tared flat-bottomed dish,and evaporate on a water bath to dryness.Dry at 105for 6hours,cool in a desiccator for 30minutes,and weigh without delay.Calculate the content,in mg per g,of alcohol-extractable matter in the test specimen.
Method 2(cold extraction method)—
Transfer about 4g of air-dried,coarsely powdered material,accurately weighed,to a glass-stoppered conical flask.Add 100mLof alcohol,insert a stopper into the flask,and macerate for 24hours,shaking frequently during the first 8hours and then allowing to stand for 18hours.Filter rapidly,taking precautions against loss of alcohol.Evaporate 25mLof the filtrate to dryness in a tared,flat-bottomed,shallow dish,and dry at 105to constant weight.Calculate the content,in mg per g,of alcohol-extractable matter in the test specimen.
Water-Soluble Extractives
Method 1(hot extraction method)—
Proceed as directed for Method 1(hot extraction method)under Alcohol-soluble Extractives,except to use water in place of alcohol.
Method 2(cold extraction method)—
Proceed as directed for Method 2(cold extraction method)under Alcohol-soluble Extractives,except to use water in place of alcohol.
Crude Fiber
Exhaust a weighed quantity of the Test Sample,representing about 2g of the drug,with ether.Add 200mLof boiling dilute sulfuric acid (1in 78)to the ether-exhausted marc,in a 500-mLflask,and connect the flask to a reflux condenser.Reflux the mixture for 30minutes,accurately timed,then filter through a linen or hardened-paper filter,and wash the residue on the filter with boiling water until the effluent washing is no longer acid.Rinse the residue back into the flask with 200mLof boiling sodium hydroxide solution,adjusted to 1.25percent by titration and free from sodium carbonate.Again reflux the mixture for 30minutes,accurately timed,then rapidly filter through a tared filter,wash the residue with boiling water until the last washing is neutral,and dry it at 110to constant weight.Incinerate the dried residue,ignite to constant weight,cool in a desiccator,and weigh the ash:the difference between the weight obtained by drying at 110and that of the ash represents the weight of the crude fiber.
NOTE—The boiling with acid and alkali should continue for 30minutes,accurately timed,from the time that the liquid (which is cooled below the boiling point by being added to the cold flask)again boils.After the solution has been brought to boiling,the heat should be turned low enough just to maintain boiling.During the boiling,the flask should be gently rotated from time to time to wash down any particles that may adhere to the walls of the flask.Aslow current of air introduced into the flask during the boiling operation aids in preventing excessive frothing.
Starch Content
Method 1—
The following is a general procedure for all reducing sugars and may be used to determine the starch content in botanical articles.
Malt Extract—
Use clean new barley malt of known efficacy,and grind just before use.Prepare malt extract just prior to use.For every 80mLof malt extract needed,digest 5g of ground malt with 100mLof water at room temperature for 2hours.[NOTE—If an electric mixer is used,stir the mixture for 20minutes.]Filter to obtain a clear extract,filtering again,if necessary,and mix the infusion well.
Test Solution—
Extract about 5g of the finely ground test specimen with five 10-mLportions of ether,using a filter that will completely retain the smallest starch granule.Allow the ether to evaporate from the residue,and wash with 250mLof aqueous alcohol solution (10in 100).Carefully wash the residue from the paper into a 500-mLbeaker with about 100mLof water.Heat to about 60(avoiding,if possible,gelatinizing starch),and allow to stand for about 1hour,stirring frequently to effect complete solution of sugars.Transfer to a wide-mouth bottle,rinse the beaker with a little warm water,and cool.Add an equal volume of alcohol,mix,and allow to stand for not less than 1hour.
Centrifuge until the precipitate is closely packed on the bottom of the bottle,and decant the supernatant.Wash the precipitate with successive 50-mLportions of alcohol solution (50in 100)by centrifuging and decanting through a suitable filter until the washings are sugar-free.[NOTE—To test for the presence of sugar,transfer a few drops of the washings to a test tube,add 3or 4drops of a 20%solution of 1-naphthol in alcohol,prepared by dissolving 200mg of 1-naphthol in 1mLof alcohol and 2mLof water.Shake the test tube well to allow uniform mixing,allow 2to 4mLof sulfuric acid to flow down the sides of the test tube,and hold the test tube upright.If sugar is present,the interface of the two liquids is colored faint to deep violet,and on shaking,the whole solution becomes blue-violet.]
Transfer the residue from the bottle and hardened filter to a beaker with about 50mLof water.Immerse the beaker in boiling water,and stir constantly for 15minutes or until all of the starch is gelatinized.Cool the beaker to 55,add 20mLof Malt Extract,and hold at this temperature for 1hour.Heat again to boiling for a few minutes,cool to 55,add 20mLof Malt Extract,and hold at this temperature for 1hour or until the residue when treated with iodine TSshows no blue tinge upon microscopic examination.Cool,dilute with water to 250mL,and filter.
General Procedure—
Transfer 200mLof the Test Solutionto a flask fitted with a reflux condenser,add 20mLof hydrochloric acid,and heat in a boiling water bath for 2½hours.Cool,nearly neutralize with sodium hydroxide TS,complete neutralization with sodium carbonate TS,dilute with water to 500mL,mix,and filter.The volume of aliquot taken depends on the starch content of the specimen under test (see Table 1).The aliquot should contain between 100and 200mg of dextrose.Transfer 50mLof the filtrate to a 400-mLalkali-resistant glass beaker,add 50mLof alkaline cupric tartarate TS,cover the beaker with a water glass,and heat.Adjust the flame in the burner so that the contents of the flask begin to boil in 4minutes and continue boiling for exactly 2minutes.Filter the hot solution at once through a sintered-glass filter.Wash the precipitate of cuprous oxide thoroughly with water at about 60,then with 10mLof alcohol,and finally with 10mLof ether.
Table 1.Determination of the Optimum Aliquot
| %of Expected Starch Content | Aliquot in mL |
| 60 | 25 |
| 50 | 35 |
| 40 | 50 |
| 30 | 50 |
| 20 | 50 |
For solutions of reducing sugars of comparatively high purity,proceed as directed under Method 1Ato determine the amount of reduced copper obtained by weighing the dried cuprous oxide.For solutions of reducing sugars containing large amounts of organic impurities,including sucrose,proceed as directed under Method 1Bto determine the amount of reduced copper obtained by titration with sodium thiosulfate.
METHOD1A—
Dry the precipitate obtained under General Procedurefor 30minutes in an oven at 110±2,cool to room temperature in a desiccator,and weigh.Refer to Table 2to find the quantity of dextrose,in mg,corresponding to the weight of cuprous oxide found.Determine the percentage of dextrose and then the content of starch by the following formula:
| Percentage of dextrose = | ||
| wt.of dextrose in mg ×0.1×500 | ||
| wt.of sample in g ×aliquot in mL | ||
| Content of starch =%dextrose ×0.9. | ||
Table 2.Calculating Dextrose (Applicable when Cu2Ois weighed directly)(Expressed in mg)
| Cuprous Oxide (Cu2O) | Dextrose (D-Glucose) | Cuprous Oxide (Cu2O) | Dextrose (D-Glucose) | Cuprous Oxide (Cu2O) | Dextrose (D-Glucose) | Cuprous Oxide (Cu2O) | Dextrose (D-Glucose) | Cuprous Oxide (Cu2O) | Dextrose (D-Glucose) | Cuprous Oxide (Cu2O) | Dextrose (D-Glucose) |
| 10 | 4.0 | 90 | 38.9 | 170 | 75.1 | 250 | 112.8 | 330 | 152.2 | 410 | 193.7 |
| 12 | 4.9 | 92 | 39.8 | 172 | 76.0 | 252 | 113.7 | 332 | 153.2 | 412 | 194.7 |
| 14 | 5.7 | 94 | 40.6 | 174 | 76.9 | 254 | 114.7 | 334 | 154.2 | 414 | 195.8 |
| 16 | 6.6 | 96 | 41.5 | 176 | 77.8 | 256 | 115.7 | 336 | 155.2 | 416 | 196.8 |
| 18 | 7.5 | 98 | 42.4 | 178 | 78.8 | 258 | 116.6 | 338 | 156.3 | 418 | 197.9 |
| 20 | 8.3 | 100 | 43.3 | 180 | 79.7 | 260 | 117.6 | 340 | 157.3 | 420 | 199.0 |
| 22 | 9.2 | 102 | 44.2 | 182 | 80.6 | 262 | 118.6 | 342 | 158.3 | 422 | 200.1 |
| 24 | 10.0 | 104 | 45.1 | 184 | 81.5 | 264 | 119.5 | 344 | 159.3 | 424 | 201.1 |
| 26 | 10.9 | 106 | 46.0 | 186 | 82.5 | 266 | 120.5 | 346 | 160.3 | 426 | 202.2 |
| 28 | 11.8 | 108 | 46.9 | 188 | 83.4 | 268 | 121.5 | 348 | 161.4 | 428 | 203.3 |
| 30 | 12.6 | 110 | 47.8 | 190 | 84.3 | 270 | 122.5 | 350 | 162.4 | 430 | 204.4 |
| 32 | 13.5 | 112 | 48.7 | 192 | 85.3 | 272 | 123.4 | 352 | 163.4 | 432 | 205.5 |
| 34 | 14.3 | 114 | 49.6 | 194 | 86.2 | 274 | 124.4 | 354 | 164.4 | 434 | 206.5 |
| 36 | 15.2 | 116 | 50.5 | 196 | 87.1 | 276 | 125.4 | 356 | 165.4 | 436 | 207.6 |
| 38 | 16.1 | 118 | 51.4 | 198 | 88.1 | 278 | 126.4 | 358 | 166.5 | 438 | 208.7 |
| 40 | 16.9 | 120 | 52.3 | 200 | 89.0 | 280 | 127.3 | 360 | 167.5 | 440 | 209.8 |
| 42 | 17.8 | 122 | 53.2 | 202 | 89.9 | 282 | 128.3 | 362 | 168.5 | 442 | 210.9 |
| 44 | 18.7 | 124 | 54.1 | 204 | 90.9 | 284 | 129.3 | 364 | 169.6 | 444 | 212.0 |
| 46 | 19.6 | 126 | 55.0 | 206 | 91.8 | 286 | 130.3 | 366 | 170.6 | 446 | 213.1 |
| 48 | 20.4 | 128 | 55.9 | 208 | 92.8 | 288 | 131.3 | 368 | 171.6 | 448 | 214.1 |
| 50 | 21.3 | 130 | 56.8 | 210 | 93.7 | 290 | 132.3 | 370 | 172.7 | 450 | 215.2 |
| 52 | 22.2 | 132 | 57.7 | 212 | 94.6 | 292 | 133.2 | 372 | 173.7 | 452 | 216.3 |
| 54 | 23.0 | 134 | 58.6 | 214 | 95.6 | 294 | 134.2 | 374 | 174.7 | 454 | 217.4 |
| 56 | 23.9 | 136 | 59.5 | 216 | 96.5 | 296 | 135.2 | 376 | 175.8 | 456 | 218.5 |
| 58 | 24.8 | 138 | 60.4 | 218 | 97.5 | 298 | 136.2 | 378 | 176.8 | 458 | 219.6 |
| 60 | 25.6 | 140 | 61.3 | 220 | 98.4 | 300 | 137.2 | 380 | 177.9 | 460 | 220.7 |
| 62 | 26.5 | 142 | 62.2 | 222 | 99.4 | 302 | 138.2 | 382 | 178.9 | 462 | 221.8 |
| 64 | 27.4 | 144 | 63.1 | 224 | 100.3 | 304 | 139.2 | 384 | 180.0 | 464 | 222.9 |
| 66 | 28.3 | 146 | 64.0 | 226 | 101.3 | 306 | 140.2 | 386 | 181.0 | 466 | 224.0 |
| 68 | 29.2 | 148 | 65.0 | 228 | 102.2 | 308 | 141.2 | 388 | 182.0 | 468 | 225.1 |
| 70 | 30.0 | 150 | 65.9 | 230 | 103.2 | 310 | 142.2 | 390 | 183.1 | 470 | 226.2 |
| 72 | 30.9 | 152 | 66.8 | 232 | 104.1 | 312 | 143.2 | 392 | 184.1 | 472 | 227.4 |
| 74 | 31.8 | 154 | 67.7 | 234 | 105.1 | 314 | 144.2 | 394 | 185.2 | 474 | 228.3 |
| 76 | 32.7 | 156 | 68.6 | 236 | 106.0 | 316 | 145.2 | 396 | 186.2 | 476 | 229.6 |
| 78 | 33.6 | 158 | 69.5 | 238 | 107.0 | 318 | 146.2 | 398 | 187.3 | 478 | 230.7 |
| 80 | 34.4 | 160 | 70.4 | 240 | 108.0 | 320 | 147.2 | 400 | 188.4 | 480 | 231.8 |
| 82 | 35.3 | 162 | 71.4 | 242 | 108.9 | 322 | 148.2 | 402 | 189.4 | 482 | 232.9 |
| 84 | 36.2 | 164 | 72.3 | 244 | 109.9 | 324 | 149.2 | 404 | 190.5 | 484 | 234.1 |
| 86 | 37.1 | 166 | 73.2 | 246 | 110.8 | 326 | 150.2 | 406 | 191.5 | 486 | 235.2 |
| 88 | 38.0 | 168 | 74.1 | 248 | 111.8 | 328 | 151.2 | 408 | 192.6 | 488 | 236.3 |
METHOD1B—
Sodium Thiosulfate Solution—
Transfer 3.9g of sodium thiosulfate,accurately weighed,to a 100-mLvolumetric flask,dissolve in and dilute with water to volume,and mix.
Potassium Iodide Solution—
Dissolve 42g of potassium iodide in 100mLof water.
Sodium Acetate Solution—
Dissolve 5.74g of sodium acetate in 10mLof water.
Copper Solution—
Transfer about 0.3g of pure electrolytic copper,accurately weighed,to a 250-mLflask,add 5mLof nitric acid to dissolve the copper,add about 25mLof water,and boil to expel red fumes.Add about 5mLof bromine TS,and boil until the bromine is completely removed.Cool,add 10mLof Sodium Acetate Solutionfollowed by 10mLof Potassium Iodide Solution,and titrate with Sodium Thiosulfate Solutionto a light yellow color.Add enough starch TSto produce a marked blue color,and continue the titration.As the endpoint nears,add 2g of potassium thiocyanate,and stir until completely dissolved.Continue titration until the precipitate is completely white.One mLof sodium thiosulfate solution is equivalent to about 10mg of copper.[NOTE—It is essential that the concentration of Potassium Iodide Solutionbe carefully regulated.If the solution contains less than 320mg of copper at the completion of titration,add 4.2to 5g of potassium iodide to make a total solution of 100mL.If greater amounts of Cu are present,add Potassium Iodide Solutionslowly from buret with constant agitation in amounts proportionately greater.]
;)
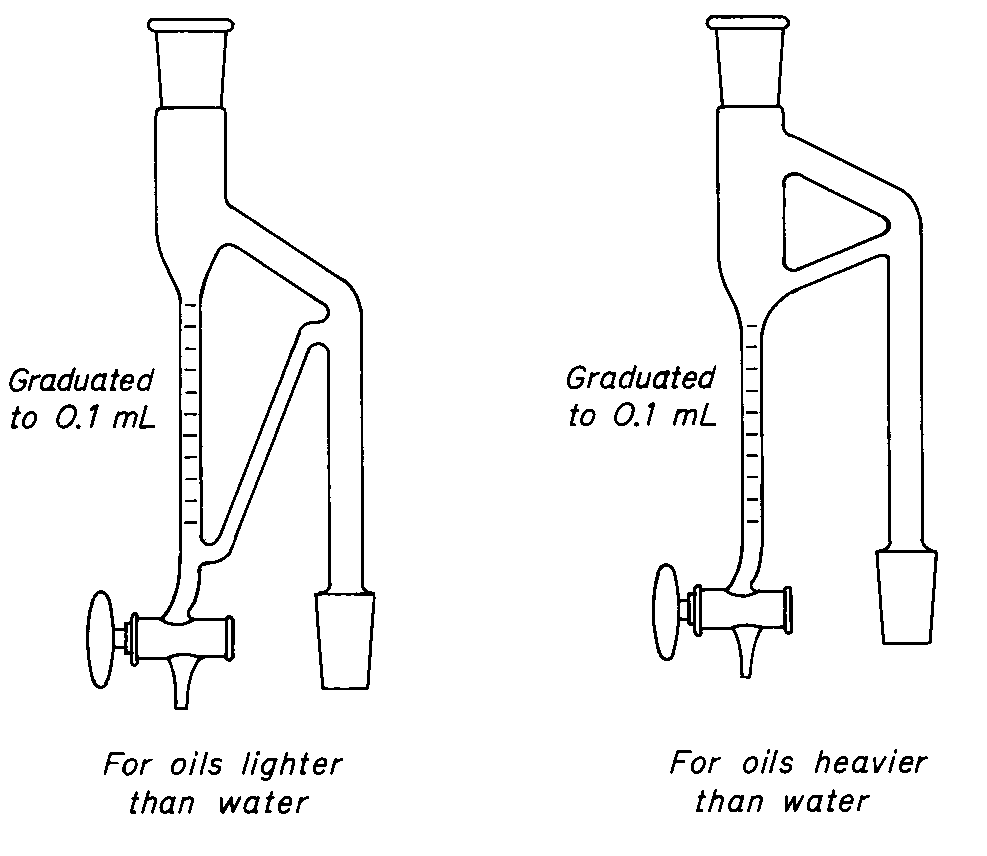
Traps for Volatile Oil Apparatus
Procedure—
Wash the precipitated cuprous oxide obtained under General Procedurewith water,cover this filter with a watch glass and dissolve the cuprous oxide with 5mLof nitric acid directed under the watch glass with a pipet.Collect the filtrate in a 250-mLflask,wash the watch glass and the filter with water.Collect all the washings in the flask.Boil the contents of the flask to expel red fumes.Add about 5mLof bromine TS,and boil until the bromine is completely removed.Cool,and proceed as directed under Copper Solutionbeginning with “add 10mLof Sodium Acetate Solution.”From the volume of Sodium Thiosulfate Solutionconsumed,obtain the weight of copper,in mg,by multiplying by 1.1259to obtain the weight,in mg,of cuprous oxide.From Table 2,find the quantity of dextrose,in mg,corresponding to the weight of cuprous oxide.The content of starch is equivalent to the weight,in mg,of dextrose obtained times 0.9.Conduct a blank determination,using 50mLof alkaline cupric tartrate TSand 50mLof Malt Extract.If the weight of the cuprous oxide so obtained exceeds 0.5mg,correct the result of the determination accordingly.[NOTE—The alkaline cupric tartrate TSdeteriorates on standing and the quantity of cuprous oxide obtained in the blank determination increases.]
Method 2—
The following method is specific for dextrose (glucose),and because of its extreme sensitivity it may account for differences noted between values obtained from the same specimen.Duplicate determinations do not vary more than 2%.
Glucoamylase Solution—
Prepare a solution of glucoamylase in water containing 30International Units (IU)per mL.Use glucoamylase obtained preferably from Rhizopus delemar.The total glucoamylase activity of the test specimen being used should be not less than 150IU.
Acetate Buffer Solution—
Dissolve 16.4g of sodium acetate in 100mLof water,add 12.0mLof glacial acetic acid,and mix.The pHof this solution is 4.8.
Phosphate Buffer—
Dissolve 3.63g of tris (hydroxymethyl)aminomethane and 5.0g of monobasic sodium phosphate in 50.0mLof water.At 37,adjust with phosphoric acid to a pHof 7.0,dilute with water to 100.0mL,and mix.[NOTE—The pHof the buffer medium is sensitive to temperature and should be adjusted to the desired pHat the temperature to be used during incubation.]
Enzyme Solution—
Dissolve 30mg of glucose oxidase (Type IIfrom Aspergillus niger),3mg of peroxidase (Type Ifrom horseradish),and 10mg of potassium ferrocyanide in 100mLof Phosphate Buffer.[NOTE—This mixture can be stored in a refrigerator for up to 10days.]
18N Sulfuric Acid—
Add slowly,while stirring,54mLof sulfuric acid to 102mLof water,allow to cool to 25,and mix.
Standard Solutions—
Dissolve an accurately weighed quantity of USP Dextrose RSin water to obtain a solution containing 1.0mg of USP Dextrose RSper mL.Quantitatively dilute a known volume of this solution with water to obtain Standard Solutions A,B,C,D,and E,having known concentrations of 10,20,25,40,and 50µg per mLof USP Dextrose RS,respectively.[NOTE—Allow 4hours for complete mutarotation before use.]
Test Solutions—
Extract about 5g of finely ground test specimen with five 25-mLportions of 80%alcohol,and filter.Remove all the alcohol from the residue by drying in an air oven at 105for about 8hours.[NOTE1—Any traces of alcohol remaining in the residue will inhibit glucoamylase.]Cool,and transfer the flask containing the dried test specimen to a desiccator.Transfer about 1g,accurately weighed,of the test specimen to a previously tared flask,add 25mLof water,and adjust with phosphoric acid to a pHbetween 5.0and 7.0,if necessary.Boil the suspension for about 3minutes,transfer the flask to an autoclave,and heat to 135for 2hours.Remove the flask from the autoclave,maintain the temperature near 55,and add 2.5mLof Acetate Buffer Solutionand sufficient water to adjust the total weight of the solution to 45±1g.Immerse the flask in a water bath maintained at 55±1,and add 5mLof Glucoamylase Solution.Continuously swirl the flask for 2hours to effect hydrolysis,filter through filter paper into a 250-mLvolumetric flask,wash quantitatively with water,and collect all the washings in the flask.Dilute the contents of the flask with water to volume,and mix.Transfer 1mLof an aliquot containing 20to 60µg of D-glucose to each of five test tubes.[NOTE2—In order to obtain the range of concentration of glucose in the hydrolysate,quantitatively dilute,if necessary,with water to volume.]Add 2mLof Enzyme Solutionto each of the five test tubes,and place the test tubes in the dark at 37±1for exactly 30minutes to develop the color.At the end of 30minutes,add 2mLof 18N Sulfuric Acidto each of the test tubes to stop the reaction,and mix.
Control Solution—
Transfer an accurately weighed quantity of about 0.4g of starch to a previously tared flask and proceed as directed under Test Solutionsbeginning with “add 25mLof water and,adjust the pHwith phosphoric acid.”
Procedure—
Concomitantly determine the absorbances of the Standard Solutionsand the Test Solutionsat the wavelength of maximum absorbance at about 540nm,with a suitable spectrophotometer,using the Control Solutionas the blank to set the instrument.Plot the absorbance values of the Standard Solutionsversus concentration,in µg per mL,of dextrose,and draw the straight line best fitting the five plotted points.From the graph so obtained,determine the concentration,C,in µg per mL,of dextrose in each of the Test Solutions,calculate the average concentration,in µg per mL,of the solution under test.The percentage of starch content in the weight of the test specimen taken by the equation is calculated by the formula:
(0.9C/106)(V1)(250/V0)(100/E)(100/W)=2.25CV1/V0EW,
in which Eis the weight,in g,of the test specimen taken;V0is the volume,in mL,of the aliquot taken from the 250-mLvolumetric flask;Wis the percentage of dry weight of the test specimen;and V1is the volume,in mL,if extra dilution is done (see Note 2under Test Solutions).[NOTE—V0is 1.0when no extra dilution is done.]
Volatile Oil Determination
Set up a round-bottom,shortneck,1-liter flask in a heating mantle set over a magnetic stirrer.Insert an egg-shaped stirring bar magnet in the flask,and attach a cold-finger condenser and an appropriate volatile oil trap of the type illustrated.
Coarsely comminute a sufficient quantity of the drug to yield from 1to 3mLof volatile oil.Small seeds,fruits,or broken leaves of herbs ordinarily do not need comminution.Very fine powders are to be avoided.If this is not possible,it may be necessary to mix them with purified sawdust or purified sand.Place a suitable quantity of the drug,accurately weighed,in the flask,and fill it one-half with water.Attach the condenser and the proper separator.Boil the contents of the flask,using a suitable amount of heat to maintain gentle boiling for 2hours,or until the volatile oil has been completely separated from the drug and no longer collects in the graduated tube of the separator.
If a proper quantity of the volatile oil has been obtained in the graduated tube of the separator,it can be read to tenths of 1mL,and the volume of volatile oil from each 100g of drug can be calculated from the weight of the drug taken.The graduations on the separator “for oils heavier than water”are so placed that oil remains below the aqueous condensate that automatically flows back into the flask.
Water Content
For unground or unpowdered drugs,prepare about 10g of the Laboratory Sampleby cutting,granulating,or shredding,so that the parts are about 3mm in thickness.Seeds or fruits smaller than 3mm should be cracked.Avoid the use of high-speed mills in preparing the sample,and exercise care that no appreciable amount of moisture is lost during the preparation and that the portion taken is representative of the Laboratory Sample.Determine the water content as directed for Procedure for Articles of Botanical Originin the Method III(Gravimetric)under Water Determination á921ñ.
Test for Aflatoxins
Caution—Aflatoxins are highly dangerous,and extreme care should be exercised in handling aflatoxin materials.
This test is provided to detect the possible presence of aflatoxins B1,B2,G1,and G2in any material of plant origin.Unless otherwise specified in the individual monograph,use the following method.
Zinc Acetate–Aluminum Chloride Reagent—
Dissolve 20g of zinc acetate and 5g of aluminum chloride in sufficient water to make 100mL.
Sodium Chloride Solution—
Dissolve 5g of sodium chloride in 50mLof water.
Test Solution 1—
Grind about 200g of plant material to a fine powder.Transfer about 50g of the powdered material,accurately weighed,to a glass-stoppered flask.Add 200mLof a mixture of methanol and water (17:3).Shake vigorously by mechanical means for not less than 30minutes,and filter.[NOTE—If the solution has interfering plant pigments,proceed as directed for Test Solution 2.]Discard the first 50mLof the filtrate,and collect the next 40-mLportion.Transfer the filtrate to a separatory funnel.Add 40mLof Sodium Chloride Solutionand 25mLof solvent hexane,and shake for 1minute.Allow the layers to separate,and transfer the lower aqueous layer to a second separatory funnel.Extract the aqueous layer in the separatory funnel twice,each time with 25mLof methylene chloride,by shaking for 1minute.Allow the layers to separate each time,separate the lower organic layer,and collect the combined organic layers in a 125-mLconical flask.Evaporate the organic solvent to dryness on a water bath.Cool the residue.If interferences exist in the residue,proceed as directed for Cleanup Procedure;otherwise,dissolve the residue obtained above in 0.2mLof a mixture of chloroform and acetonitrile (9.8:0.2),and shake by mechanical means if necessary.
Test Solution 2—
Collect 100mLof the filtrate from the start of the flow,and transfer to a 250-mLbeaker.Add 20mLof Zinc Acetate–Aluminum Chloride Reagentand 80mLof water.Stir,and allow to stand for 5minutes.Add 5g of a suitable filtering aid,such as diatomaceous earth,mix,and filter.Discard the first 50mLof the filtrate,and collect the next 80-mLportion.Proceed as directed for Test Solution 1,beginning with “Transfer the filtrate to a separatory funnel.”
Cleanup Procedure—
Place a medium-porosity sintered-glass disk or a glass wool plug at the bottom of a 10-mm ×300-mm chromatographic tube.Prepare a slurry of 2g of silica gel with a mixture of ethyl ether and solvent hexane (3:1),pour the slurry into the column,and wash with 5mLof the same solvent mixture.Allow the absorbent to settle,and add to the top of the column a layer of 1.5g of anhydrous sodium sulfate.Dissolve the residue obtained above in 3mLof methylene chloride,and transfer it to the column.Rinse the flask twice with 1-mLportions of methylene chloride,transfer the rinses to the column,and elute at a rate not greater than 1mLper minute.Add successively to the column 3mLof solvent hexane,3mLof ethyl ether,and 3mLof methylene chloride;elute at a rate not greater than 3mLper minute;and discard the eluates.Add to the column 6mLof a mixture of methylene chloride and acetone (9:1),and elute at a rate not greater than 1mLper minute,preferably without the aid of vacuum.Collect this eluate in a small vial,add a boiling chip if necessary,and evaporate to dryness on a water bath.Dissolve the residue in 0.2mLof a mixture of chloroform and acetonitrile (9.8:0.2),and shake by mechanical means if necessary.
Aflatoxin Solution—
[Caution—Aflatoxins are highly toxic.Handle with care.
]Dissolve accurately weighed quantities of aflatoxin B1,aflatoxin B2,aflatoxin G1,and aflatoxin G2in a mixture of chloroform and acetonitrile (9.8:0.2)to obtain a solution having concentrations of 0.5µg per mLeach of aflatoxin B1and aflatoxin G1,and 0.1µg per mLeach of aflatoxin B2and aflatoxin G2.
Procedure—
Separately apply 2.5µL,5µL,7.5µL,and 10µLof the Aflatoxin Solutionand three 10-µLapplications of either Test Solution 1or Test Solution 2to a suitable thin-layer chromatographic plate (see Chromatography á621ñ)coated with a 0.25-mm layer of chromatographic silica gel mixture.Superimpose 5µLof the Aflatoxin Solutionon one of the three 10-µLapplications of the Test Solution.Allow the spots to dry,and develop the chromatogram in an unsaturated chamber containing a solvent system consisting of a mixture of chloroform,acetone,and isopropyl alcohol (85:10:5)until the solvent front has moved not less than 15cm from the origin.Remove the plate from the developing chamber,mark the solvent front,and allow the plate to air-dry.Locate the spots on the plate by examination under UVlight at 365nm:the four applications of the Aflatoxin Solutionappear as four clearly separated blue fluorescent spots;the spot obtained from the Test Solutionthat was superimposed on the Aflatoxin Solutionis no more intense than that of the corresponding Aflatoxin Solution;and no spot from any of the other Test Solutionscorresponds to any of the spots obtained from the applications of the Aflatoxin Solution.If any spot of aflatoxins is obtained in the Test Solution,match the position of each fluorescent spot of the Test Solutionwith those of the Aflatoxin Solutionto identify the type of aflatoxin present.The intensity of the aflatoxin spot,if present in the Test Solution,when compared with that of the corresponding aflatoxin in the Aflatoxin Solutionwill give an approximate concentration of aflatoxin in the Test Solution.