MANUFACTURING OF GENE THERAPY PRODUCTS
Introduction
The principles applicable to the production of pharmaceutical or biological products are also relevant to the production of gene therapy vectors for therapeutic use in humans.The same CGMPrequirements can be applied to determine product consistency,process validation,raw material qualification,and compliance of the manufacturing facilities.Manufacturers will face development issues such as scalability,yield,cost efficiency,and product stability.
Most gene therapy vectors have been produced only in relatively small batches necessary to meet the needs of early clinical trials in small numbers of patients.However,areas of rapid progress are large-scale production of vectors,vector purification,and suitable analytical techniques.This section focuses on issues involved with designing vectors for gene therapy and choosing a production technology;it does not focus on specific production technologies.
Design Considerations for Gene Vectors
TYPES OF VECTORS
Atypical gene therapy vector is composed of (1)the vector backbone,viral or plasmid,(2)a promoter,(3)the therapeutic gene of interest,including introns,and (4)a polyadenylation signal.Murine and human retroviruses,adenoviruses,parvoviruses such as adeno-associated virus (AAV),herpes viruses,poxviruses,toga viruses,nonviral plasmid therapy systems,and synthetic antisense-oligonucleotide therapy systems are being developed for gene therapy applications.The properties of these vectors (see Table 5)differ greatly in terms of their capacity to deliver genes to cells.Some viral vectors preferentially target dividing cells while others are capable of transducing both dividing and nondividing cells.There are significant variations in transgene capacity,meaning that there are limitations on the size of the foreign DNAfragment that can be incorporated into the recombinant genome.The ideal gene therapy vectorhas often been described as one capable of efficient transduction,targeted delivery,and controlled gene expression.The level,timing,and duration of gene expression required will depend on the clinical indication.Low-level,long-term gene expression is thought to be required for some diseases including adenosine deaminase (ADA)deficiency or type Aand type Bhemophilia.High-level,short-term expression may be more appropriate for cancer when genes that induce apoptosis are used,or for cardiovascular disease where preventing hyperproliferation of smooth-muscle cells may impede restenosis of saphenous vein grafts.
Table 5.Types of Gene Vectors
| FAMILY | RETROVIRIDAE | ADENOVIRIDAE | VIRAL PARVOVIRIDAE | HERPES- VIRIDAE |
TOGAVIRIDAE | POXVIRIDAE | NONVIRAL | |
| Example Species | Murine Leukemia Virus | HIV | Adenovirus | AAV | Herpes Simplex Virus | Sindbis | Poxvirus (Vaccinia) |
Plasmid derived |
|
Vector Characteristics |
||||||||
| Insert size limit | 8kb | 8kb | 4.3to 34kb | 4to 5kb | 40to 150kb | 5kb | 25to 50kb | 12kb |
| Chromosome integration | Yes | Yes | No;episomal | Can be integrated or episomal | Can be integrated or episomal | No | No | Yes,but at very low frequency |
| Therapeutic protein expression | Stable | Stable | Stable or transient | Stable | Stable or transient | Transient | Transient | Stable or transient |
| Vector localization | Nucleus | Nucleus | Nucleus | Nucleus | Nucleus | Cytoplasm | Cytoplasm | Nucleus |
| Types of cells transduced | Dividing only | Dividing and quiescent |
Dividing and quiescent |
Dividing and quiescent |
Dividing and quiescent | Dividing and quiescent |
Dividing and quiescent |
Dividing and quiescent |
| Efficiency of gene transfer | High | High | High | High | High | High | High | Low |
| Expression of viral proteins | No | No | Yes,unless viral genes deleted | No | Yes | Yes | Yes | No |
| Other | Tropism can be altered by pseudotyping |
Can be used as a plasmid therapy system | ||||||
VECTOR DESIGN CRITERIA
Vectors are designed and selected for disease states on the basis of the following criteria:
-
capacity to accommodate the DNAfor the therapeutic gene and its transcription cassette
-
host–vector interactions,both cellular and humoral
-
capacity to target intended cells
-
control of therapeutic gene expression
-
vector replication status
-
capacity for integration into chromosomes of target cells.
Selection of the route of administration and manipulation of the total dose of vector are strategies that can be used to compensate for some features of specific vector systems.The design and selection of a vector system include the evaluation of the disease of interest.
Additionally,there are advantages and disadvantages for the manufacture of each of the different vector systems.Production consistency favors those systems with well-defined fermentation or culture systems,such as plasmid,retroviral,or adenoviral vectors,or chemically defined systems,such as synthetic antisense-oligonucleotide systems.For those viral vector systems that require helper functions (see below),a rationally engineered cell line can overcome the scalability and consistency limitations of cotransfections.Engineered cell lines can also eliminate the possibility of replication-competent recombinant virus appearing in viral culture.Use of a cell line that is adapted to suspension culture can affect scalability and cost efficiency.
TARGETING TRANSDUCTION
To be effective,a vector must first find and transduce its target cell.Viruses have a natural host range that is strongly influenced by the expression levels of specific cell-surface receptors in target tissues,the cell cycle status of the target cells,and the route of administration.Integrins are a class of cell-adhesion receptors known to interact with either the penton base or the fiber protein of adenoviruses.The Coxsackie and adenovirus receptor (CAR)is also known to interact with adenoviruses.However,the expression levels of integrins and of CARvary according to tissue type,affecting the transduction efficiency of adenoviral vectors.Amphotropic retroviruses infect cells via a sodium-dependent phosphate transporter molecule that is expressed at a detectable level in every human cell type.
The host and tissue range can be modified or targeted by a variety of approaches.Retroviruses,and lentiviruses,in particular,encode an envelope protein that mediates virus binding and entry via a specific host-cell receptor.Envelope proteins from one retrovirus may be interchanged with a protein from another retrovirus or a protein,such as the vesicular stomatitis virus glycoprotein from an entirely different virus.This process is referred to as pseudotyping.Viral protein coats may be modified in several ways.By engineering the fiber and knob of adenovirus,it is possible to change the intrinsic integrin specificity.Similarly,viral coat proteins can be chemically modified for ligand-mediated receptor targeting.It is feasible to create ligand–plasmid fusion molecules for receptor-mediated targeting of nonviral vectors.Some lipid formulations for nonviral vectors incorporate antibody Fab fragments or ligands to target plasmid delivery.
With respect to cell cycling,adenoviruses easily infect both quiescent and rapidly dividing cells,while murine leukemia virus-based retroviral vectors are efficient only when transducing rapidly dividing cells.Lentiviral vectors can infect quiescent cells,including cells of neuronal origin.In general,nonviral vectors have lower transduction efficiencies than viral vectors.Transduction efficiencies of nonviral vectors are strongly influenced by the formulation used and route of administration.
IMPACT OF HUMORAL IMMUNE SYSTEM
Regardless of the route of administration,the intended target cell,and the dose,the vector is likely to encounter some component of the immune system as it moves toward the target cell.For viral vectors,the humoral (antibody-based)immune system cannot readily distinguish between wild-type viral infections and recombinant viral vectors because the humoral response is directed against proteins contained in the viral coat or package.Protein-containing formulations of nonviral vectors can also elicit a humoral immune response.Either specific or cross-reacting humoral responses may pre-exist or they may be elicited during dosing,and the antibody response may vary in its capacity to diminish gene transduction in individual patients.It is possible to compensate for the neutralizing activity of the antibodies by increasing the vector dose or by altering the dosing interval to coincide with periods of low antibody titer.Because neutralizing capacity is frequently enhanced upon multiple dosing,effective dosing by repeated administration may be problematic.This issue is generally avoided by the use of nonviral systems.Alternatively,viral vectors can be engineered to evade the immune system.For adenoviral vectors,one approach involves increasing expression of specific viral genes that allow the virus to evade the host's humoral response.
IMPACT OF CELLULAR IMMUNE RESPONSES
Once protein expression is under way,cellular immune responses can lead to a rapid removal of both viral and nonviral vector-transduced cells from the body and a decrease in therapeutic effectiveness.Although protein synthesis is not required for cellular immune responses to viral vector envelope proteins,de novo synthesis of viral genes can exacerbate host-cellular responses.To reduce potential cellular responses,viral vectors have been designed with specific backbone deletions to eliminate the expression of viral structural genes.Examples of such vectors include the E1-and E4-deleted adenoviruses,the adenoviruses and herpesviruses in which all viral genes have been deleted (gutless)or are helper dependent,and the recombinant adeno-associated viral vectors.Certain plasmid sequences expecially those with the CpGmotif,can elicit a strong cellular immune response.
ANTIGENICITY OF GENE PRODUCT
The therapeutic gene product may also be antigenic.When proteins that are retained in the target cell are used,cellular responses may eliminate the target cell.In some cases this is the desired therapeutic effect,particularly in the antigen-based immunotherapy for cancer or a viral disease.However,if sustained protein expression is required,the cellular immune response may decrease the effectiveness of the therapy or eliminate it entirely.The antigenicity of the therapeutic gene may reflect a variety of experimental conditions.If a gene such as the cystic fibrosis transmembrane conductance regulator (CFTR)is truncated to fit within a chosen vector,this modification may result in creation of a distinct antigen.By using the gene that encodes thymidine kinase derived from the herpes simplex virus (HSV),a foreign protein is introduced into a human subject and thus it can function as an antigen.Any patient with a monogenic deficiency disorder is at risk for lack of tolerance to the normal protein that is defective or absent in the disease state (for example,dystrophin in Duchenne muscular dystrophy).
COMPLEMENT INACTIVATION
Retroviral vectors are also subject to another host-defense mechanism—the complement component of the immune system.Retroviral vectors are reported to be rapidly inactivated by complement in sera from primates,but not from lower mammals.In considering the replication cycle of retroviruses,it is known that glycosylation epitopes are derived from the host cell during the budding process.Because many retroviral vectors used in gene therapy are murine in origin and have been grown in mouse packaging cell lines,they will have envelopes containing mouse glycoproteins.When retroviral vectors are made in human cells,they are substantially more resistant to human complement.It is reasonable to assume that the mechanism of resistance involves incorporation of natural human cell-membrane complement control proteins that have been incorporated into the vector envelope during the budding stage of particle assembly.
VECTOR LOCALIZATION WITHIN THE TARGET CELL
Once the vector reaches the target cell,several factors can affect the level and duration of therapeutic gene expression,and these factors dictate the choice of an appropriate vector system for a specific clinical indication.The localization of the vector genome within the cell,the strength of the gene expression control elements,the stability of the message,and the stability of the translated protein will all affect the therapeutic impact.Alphavirus-based vectors,such as those derived from Sindbis or Semliki Forest virus,reside in the cytoplasm and typically exhibit a very high level of gene expression.Retroviral,adenoviral,and other viral vectors have advantages in gene delivery with their natural mechanisms for nuclear delivery of the therapeutic gene and reasonable levels of gene expression from viral or other promoters.Nonviral plasmid vectors are episomal and are often susceptible to DNAdegradation when they are shunted into cell endosomes.However,some nonviral systems incorporate nuclear targeting signals as a means of increasing therapeutic gene-transcription efficiency.
TISSUE-SPECIFIC PROMOTERS
Another means of controlling gene expression is to incorporate tissue-specific promoters to stimulate or to restrict expression of the therapeutic gene.Drug-responsive promoters are being used to control gene expression.Rapamycin,mifepristone,or the tetracycline onsystems have been used to repress gene expression.This type of regulation may be required for certain proteins,such as erythropoietin,where constitutive expression may produce toxicity.
IMPACT OF REPLICATION STATUS OF VECTOR
Replication status is another important consideration for vector design and selection.Viral vectors are most frequently constructed to be incompetent or replication-defective in order to limit uncontrolled vector spread and pathogenicity.However,when effective therapy requires infection of virtually all the target cells,replication can be engineered to be conditional when specific viral gene interactions are matched with intracellular pathway targets.When these targets are defective or missing,such as in cancer cells,the virus can replicate,but when the target cell is functioning normally,viral replication is repressed.One of the risks inherent in the use of conditionally replicating viral vectors is that the growth of the virus is not absolutely restricted to a single cell type,that is,the system may be leaky.As compensation,the susceptible target cells may be efficiently transduced at a dose that is significantly lower than that necessary for nontarget cells.
Nonviral vectors are normally designed as nonreplicating systems,but some groups are experimenting with replicating nonviral plasmids to increase gene expression levels given the low transduction efficiency of most nonviral systems and to increase the duration of gene expression.Additional preclinical studies are needed to establish the safety of these systems.Artificial chromosomes have also been designed to take advantage of normal mechanisms for retaining gene expression in rapidly dividing target cells.
VECTOR INTEGRATION
The duration of gene expression is also a function of the stability of the vector genome.Retroviral vectors can stably integrate into the host-cell genome.Adenoviruses do not integrate because their DNAremains episomal.Recombinant adeno-associated virus (AAV)vectors integrate,but because the repgenes responsible for site-specific integration are normally excluded from the construct in order to increase the vector-packaging capacity,integration is not site-specific as it is for wild-type AAV.Nonviral plasmid DNAdoes not integrate efficiently.However,stable episomes have been observed in certain cell types,such as muscle cells.Site-specific integration can be a desirable feature for vectors intended to correct genetic disorders.Although it is not currently possible,the control of the site of integration is desirable in order to prevent insertional mutagenesis.Insertional mutagenesis has the potential to kill a cell,if a critically functioning gene is inactivated,or to predispose a cell to malignant transformation,if a tumor-suppressor gene is inactivated.
The success of any gene therapy product is dependent on the relationship between the vector-delivery system and the requirements of the disease application in terms of the site,level,and duration of therapeutic gene expression.It is unlikely that there will ever be a universal vector,and the challenge is in fitting the vector to the disease.
Manufacturing and Purification Strategies
VECTOR CONSTRUCTION
Viral and nonviral gene-transfer vectors are constructed by using standard molecular biology protocols.For viral vectors,the vector backbone consists of viral RNAor DNAsequences from which the regions encoding viral structural genes or the regions required for replication have been deleted.The deleted region of the vector is usually modified with specific restriction endonuclease sites used to allow insertion of the gene of interest.For nonviral vectors,the plasmid DNAbackbone contains multiple restriction sites for cloning and the bacterial elements necessary for plasmid production.Vector backbones can accommodate single or multiple gene inserts depending on the maximum amount of sequence they can carry.The promoter that facilitates transcription of the gene insert can be a related viral promoter,such as murine leukemia virus long terminal repeat (MuLV LTR),or a heterologous promoter that is either tissue-specific,such as alpha crystalline promoter (of the eye),or constitutive,such as cytomegalovirus (CMV).For example,in a retroviral vector construct containing two gene inserts,transcription of one is regulated from the 5¢-LTR-promoter sequence,while a second gene insert can be linked to an internal heterologous promoter from Simian virus 40(SV40).The complementary DNA(cDNA)containing the therapeutic gene of interest,including its introns,is excised from its source by using restriction enzymes and is inserted at the multiple cloning site of the gene-transfer vector.The polyadenylation signal can be derived from multiple sources such as the SV40virus or human growth hormone.Characterization and testing of gene therapy vectors are described under Analytical Methodologies.
HELPER FUNCTION SYSTEMS
Recombinant viral vectors are most often modified to be replication-defective,a condition created by deletion or modification of the viral genes needed for replication and production of infectious virus.As a result,viral vectors require help to produce infectious vector particles.Helper functions are often provided by packaging cell lines to deliver the necessary viral element from a source outside of the gene of interest (in trans).Packaging cell lines should be designed to minimize the risk of production of RCVthrough recombination between the vector and the packaging elements.
Plasmids encoding the necessary elements are introduced into the packaging cell by standard methods such as calcium phosphate–mediated transfection or electroporation.If multiple trans-acting elements are needed,these elements are introduced on separate plasmids in order to increase the number of recombination events needed to form a wild-type viral genome,thus decreasing the frequency of the event.An additional approach to eliminate production of RCVis the elimination of common sequences between the packaging cell plasmids and the gene therapy vector.
Stable packaging cell lines should be selected and clonal MCBs prepared.In retroviral vector production systems,typically the pro-viral form of the retroviral vector is stably incorporated into the packaging cell,resulting in what is referred to as the producer cell line.Astable,banked packaging cell–producer line will lead to consistency in production and control of adventitious agent contamination.Alternatively,the system can be transient,with the packaging plasmids transfected along with the gene therapy vector for each round of vector production.However,a transient transfection system is less efficient and limited in scalability.
Typical helper function systems are as follows:
-
Retroviral Vector Systems—The murine fibroblast cell line NIH3T3has been the basis for several packaging cell lines.The gag,pol,and envfunctions can be colocated on a single plasmid (PA317)or placed on individual plasmids (psi-CRIP).This increases the number of recombination events required to produce an RCV.The human embryonic kidney cell line 293has been modified to be a packaging cell line for retroviruses,because use of a human cell line allows production of a retroviral vector that is not affected by the human complement system.
-
Adenoviral Vector Systems—HEK293cells are widely used to supply the E1function necessary for efficient adenoviral replication that is deleted from first-generation adenoviral vectors.Other complementing cell lines,such as E1-modified A549cells (human lung carcinoma)and the PER.C6cell line (human embryonic retinoblast),have also been created to supply E1or other missing functions.The latter contains the E1region under the control of a phosphoglycerate kinase (PGK)promoter and has no flanking adenoviral sequences in order to eliminate production of replication-competent adenovirus (RCA).
-
AAV Vector Systems—These systems classically use adenovirus-infected human 293cell lines transiently transfected with AAVhelper plasmid containing the repand capgenes,which are required for AAVreplication and capsid formation,respectively,and which are deleted from the AAVvector.The HeLa cell line (from human uterine cervical carcinoma)has also been used as a transient production system.More recently,both of these cell lines have been used to establish stably transfected packaging cell lines that express repand capgenes and in some cases express the adenoviral functions needed for AAVreplication when repand capare present (E1a,E1b,E2a,E4,and VA RNA).
-
Gutless Adenoviruses—The manufacturing systems for gutless adenoviruses are similar to classical AAVvector manufacturing systems in that human 293cells are transiently transfected with helper plasmid containing required adenoviral functions.
VIRAL GENE THERAPY VECTORS
Retrovirus and adenovirus have classically been produced on the laboratory scale by using traditional cultivation methods for anchorage-and serum-dependent cell lines,employing flasks,trays,and roller bottles.Initially,gene therapy vectors were produced by using these exact methods because large volumes of product were not required for early clinical studies.Cell bank systems are used as the source of cells and virus banks as the source of virus for clinical production.In most cases,supernatant is collected,clarified,and stored frozen in bags at -70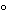 .In many early clinical trials unpurified supernatant has been used for ex vivo gene transfer.
.In many early clinical trials unpurified supernatant has been used for ex vivo gene transfer.
More recently,larger-scale upstream production methods have been reported including suspension,bioreactor,and fixed-bed or microcarrier culture methods.Some groups have reported adapting their process cells to serum-free culture conditions.Cells are harvested and lysed or supernatant collected.The harvest is clarified and purified to remove host-cell debris,host-cell DNA,and other process-derived contaminants.
Traditionally,viruses are purified by gradient ultracentrifugation,but this is time-consuming and unsuitable for larger-scale production purposes.The selection of downstream process steps and their sequence is determined by the nature of the virus itself and the upstream process used for manufacturing the virus.As processes are being developed for the manufacture of gene therapy vectors,many different purification steps have been reported.These include ion-exchange and sulfonated-cellulose chromatography,zinc ion affinity chromatography,size-exclusion chromatography,and DNase or other nuclease treatments.AAVproduction and lentiviral production are complicated by a need for transient transfection or cotransfection of plasmid or helper virus.These processes have so far required anchorage-dependent cell lines that are difficult to scale up.The development of stably transfected cell lines would allow large-scale production.
PLASMID VECTORS
Plasmids are double-stranded,circular DNAmolecules that exist in bacteria as extrachromosomal,self-replicating molecules.They have been modified to serve as cloning systems,to contain multiple restriction endonuclease recognition sites for insertion of the cloned transgene,and to contain selectable genetic markers for identification of cells that carry the recombinant vector.Plasmid-based nonviral vectors are frequently used as gene delivery systems for both in vivo and ex vivo gene therapies.They are in the form of naked DNAor complexed with lipids or other agents that facilitate transfer across the cell membrane and delivery to the cell nucleus without degradation.An advantage of a plasmid-vector system is the efficient production of large quantities of the vector that is easily characterized and involves no risk of contamination with the RCV.
Nonviral vectors are typically produced by using an Escherichia colibacterial system.Plasmids are transfected into Escherichia coli,and an appropriate single bacterial colony is selected and expanded to create an MCB.After reselection of a colony from a bacterial plate inoculated from the MCB,plasmid DNAis isolated from cultures that can range in size from 1Lon a laboratory scale to hundreds of Lin bacterial fermenters.Plasmid DNAcan be purified by several methods including affinity or ion-exchange chromatography and cesium chloride–ethidium bromide density gradients.Cesium chloride–ethidium bromide density gradients are not recommended for production of clinical-grade material.
OLIGONUCLEOTIDE VECTORS
Antisense oligonucleotides are manufactured by synthetic chemistry procedures.Currently,the method of choice is solid-phase phosphoramidite chemistry.Synthesis is linear,rather than convergent,and a high level of efficiency must be maintained at each synthesis step.This is accomplished by using molar excesses of highly pure raw materials to drive the reaction kinetics towards completion.Synthetic oligonucleotide manufacturing may require metric-ton quantities of nucleoside phosphoramidites and other compounds such as activator and sulfur-transfer reagents for commercial-scale manufacturing.An issue for oligonucleotide manufacturing is that during the preparation of raw materials and the synthesis of oligonucleotides,precaution must be taken regarding moisture,because moisture is detrimental to both yield and purity.Purification of the single-strand oligonucleotide product requires removal of residual solvents and synthetic strand by-products.Nevertheless,current oligonucleotide-manufacturing technology is readily scalable and cost-efficient,and it results in products with purity levels similar to those of classical small-molecule pharmaceuticals.
FORMULATION OF GENE THERAPY PRODUCTS
Final formulations for vector products are still in early development.So far,mannitol,sucrose,lipids,polymers,and serum albumin have been utilized as stabilizers.Aseptic filling of large numbers of vials,using classical manufacturing processes,may be problematic.For example,some viral vectors are thermally sensitive and storage at ultra-low temperatures is often required.Progress is being made in both viral and nonviral vector lyophilization and in the use of stabilizers for liquid formulations.