Atropine
;)
Benzeneacetic acid,a-(hydroxymethyl)-8-methyl-8-azabicyclo[3.2.1]oct-3-yl ester,endo-(±)-.
1aH,5aH-Tropan-3a-ol (±)-tropate (ester) [51-55-8].
»Atropine contains not less than 99.0percent and not more than 100.5percent of C17H23NO3,calculated on the anhydrous basis.
Packaging and storage—
Preserve in tight,light-resistant containers.
Identification—
A:
Dissolve 30mg of Atropine and 36mg of USP Atropine Sulfate RSin individual 60-mLseparators with the aid of 5-mLportions of water.To each separator add 1.5mLof 1Nsodium hydroxide solution and 10mLof chloroform.Shake for 1minute,allow the layers to separate,and filter the chloroform extracts through separate filters of about 2g of anhydrous granular sodium sulfate supported on pledgets of glass wool.Extract each aqueous layer with two additional 10-mLportions of chloroform,filtering and combining with the respective main extracts.Evaporate the chloroform solutions under reduced pressure to dryness,and dissolve each residue in 10mLof carbon disulfide:the IRabsorption spectrum,determined in a 1-mm cell,of the solution obtained from the test specimen exhibits maxima only at the same wavelengths as that of the solution obtained from the Reference Standard.
B:
To a 1in 50solution in 3Nhydrochloric acid add gold chloride TS:a lusterless precipitate is formed (distinction from hyoscyamine,which,similarly treated,yields a lustrous precipitate).
Angular rotation á781Añ:
the angular rotation of this solution,a 200-mm tube being used,is between -0.70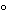 and +0.05(limit of hyoscyamine).
and +0.05(limit of hyoscyamine).
Test solution—
Dissolve 1g,previously dried at 105for 1hour,in sufficient 50%alcohol (w/w)to obtain a volume of 20mLat 25.
Water,Method Iá921ñ:
not more than 0.2%.
Residue on ignition á281ñ:
not more than 0.1%.
Readily carbonizable substances á271ñ—
Dissolve 200mg in 5mLof 2Nsulfuric acid:the solution has no more color than Matching Fluid A,and the solution is colored no more than light yellow upon the addition of 0.2mLof nitric acid.
Limit of foreign alkaloids and other impurities—
Prepare a solution of Atropine in methanol containing 20mg per mL,and,by quantitative dilution of a portion of this solution with methanol,prepare a second solution of Atropine containing 1mg per mL.Apply 25µLof the first (20mg per mL)Atropine solution,1µLof the second (1mg per mL)Atropine solution,and 5µLof a methanol solution of USP Atropine Sulfate RScontaining 24mg per mLto a suitable thin-layer chromatographic plate (see Chromatography á621ñ)coated with a 0.5-mm layer of chromatographic silica gel.Allow the spots to dry,and develop the chromatogram in a solvent system consisting of a mixture of chloroform,acetone,and diethylamine (5:4:1)until the solvent front has moved about three-fourths of the length of the plate.Remove the plate from the developing chamber,mark the solvent front,and allow the solvent to evaporate.Locate the spots on the plate by spraying with potassium iodoplatinate TS:the RFvalue of the principal spot obtained from each test solution corresponds to that obtained from the Reference Standard solution;no secondary spot obtained from the first Atropine solution exhibits intensity equal to or greater than the principal spot obtained from the second Atropine solution (0.2%).
Organic volatile impurities,Method Vá467ñ:
meets the requirements.
Solvent—
Use dimethyl sulfoxide.
Assay—
Dissolve about 400mg of Atropine,accurately weighed,in 50mLof glacial acetic acid,add 1drop of crystal violet TS,and titrate with 0.1Nperchloric acid VSto a green endpoint.Perform a blank determination,and make any necessary correction.Each mLof 0.1Nperchloric acid is equivalent to 28.94mg of C17H23NO3.
Auxiliary Information—
Staff Liaison:Ravi Ravichandran,Ph.D.,Senior Scientist
Expert Committee:(PA3)Pharmaceutical Analysis 3
USP28–NF23Page 200
Phone Number:1-301-816-8330