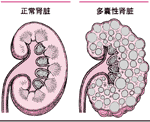
第126节 代谢性和先天性肾脏疾病 肾脏疾病可能源于解剖或代谢异常,很多是遗传性并在出生时就存在(先天性)。
. 症状 巴特尔综合征的患儿生长缓慢,表现营养不良,可有肌无力和极度口渴,多尿,智力发育迟缓。 血中氯化钠浓度和水分降低,机体代偿性产生较多的醛固酮和肾素,这些激素则降低血钾浓度(见第137节)。 . 诊断和治疗 根据症状医生作出巴特尔综合征的可疑诊断,实验室检查结果发现血钾和激素浓度异常则支持本病诊断。
口服钾盐补充剂和减少尿排钾的药物,如螺内酯(也阻断醛固酮的作用)、氨苯蝶啶、阿米洛利、普萘洛尔或吲哚美辛可预防本病的许多并发 症。为代偿过多水分丢失,则应足量饮水。